AI 如何影响科学?[译]
Michael Nielsen,
Astera Institute
到目前为止,AI 在科学领域最大的成就莫过于 AlphaFold 2 系统。这是一个深度学习系统,它在解决一个基础科学难题上取得了巨大进步:从蛋白质的氨基酸序列预测其三维结构。这一重大突破促进了分子生物学领域深度学习革命的持续进行。这不仅对分子生物学家来说意义重大,而且我认为它对整个科学领域都极具意义,因为它是一个具体的实例,展示了人工智能如何影响科学发现。在这个简短的报告中,我将探讨几个问题:如何验证这样的系统?它们能否帮助我们找到人类科学家可以学习的普遍原则?一个优秀的理论或解释应该包含哪些内容?本次讲座主要关注现有成果和近期发展,而非长远未来。然而,我希望它能帮助我们更好地理解未来的长期走向,以及 AI 系统所带来的好处和风险。
这是在 2023 年 5 月于华盛顿特区举办的Metascience 2023 会议上作的演讲。
2020 年,深度学习系统 AlphaFold 21 在生物学界引起了轰动,它被证明能够常规地进行几乎具有原子级精度的蛋白质结构预测。也就是说,AlphaFold 可以仅通过蛋白质的氨基酸线性序列,准确预测蛋白质中原子的位置。这些成果不是挑选出来的,而是在与一百多个其他建模团队的公开竞争中获得的。在某些情况下,AlphaFold2 的预测甚至超过了实验精度,使得现有的实验结果得到了重新评估和改进。
虽然 AlphaFold 非常引人注目,但它还远非完善:它实际上是通向新时代的一座桥梁,开辟了众多科学和元科学(metascience)问题。这些问题包括:我们期望一个好理论或解释能提供什么;如何验证这种理解;以及我们人类能从这些系统中学到什么。最重要的问题是:AI 系统是否以及如何可能影响科学整体的进展,作为一种系统性的干预。在我的演讲中,我将把 AlphaFold 作为一个具体的例子,来展示 AI 如何可以跨学科使用。因此,即便你对蛋白质或生物学不感兴趣,作为元科学家(metascientist)与 AlphaFold 及其后续系统的互动仍然非常有价值。
这次演讲是一次概述。我不是分子生物学家(molecular biologist),如果在这方面有任何错误,我在此表示歉意。尽管如此,学习关于蛋白质的知识仍然是一件愉快且富有启发性的事情。请允许我用几分钟时间为你们这些非生物学家(就像我一样)回顾一些基础知识。分子生物学(molecular biology)就像走进一个充满了奇妙而多样化机器的巨大工作室。像 UniProt 这样的大型数据库记录了数亿种蛋白质的氨基酸序列。比如,你可能听说过运动蛋白(kinesin),它们在细胞内部帮助运输大分子物质。还有血红蛋白(haemoglobin),它在你的血液中运输氧气,助力你的新陈代谢。还有绿色荧光蛋白(green fluorescent protein),在紫外光照射下会发出绿光,用于标记和追踪其他生物分子。所有这些以及更多的分子机器,都是通过自然选择这个严苛的筛选过程创造和分类的。每一种机器都足以成为终生研究的对象。举个例子,仅运动蛋白(kinesin)超家族就有成千上万的研究论文,但我们对它的了解仍在起步阶段。然而,尽管这些生物机器的多样性令人赞叹,我们并不是先验地(a priori)知道这些机器的功能或它们的工作方式。我们没有操作手册,正努力去揭开它们的秘密。
事实上,我们已知的数亿种蛋白质中的大多数,我们能够直接确定的,只是它们的基本结构图纸。通过基因组测序,我们可以找出蛋白质由哪些氨基酸按顺序排列而成,这个过程成本极低。然而,蛋白质实际上是纳米级的微小三维结构,这使得直接观察它们非常困难。要确定一个蛋白质的三维结构,通常需要花费数月的时间,常用的方法包括 X 射线晶体学、冷冻电镜技术或核磁共振(NMR)。
这种差异非常关键,因为了解蛋白质的形状对于解答以下问题至关重要:
- 抗体蛋白可以绑定哪些抗原,以促进免疫反应?
- 蛋白质能携带哪些物质?(例如,血红蛋白如何携带氧气)
- 蛋白质如何组成更大的复合结构?(例如,核糖体的形成)
虽然了解蛋白质的形状并不能完全揭示其功能,但这是理解蛋白质功能和作用机理的基础。
理想情况下,我们希望能够仅通过分析氨基酸序列来推断出蛋白质的形状。化学家和生物学家普遍认为这通常是可行的。自 20 世纪 70 年代起,科学家就开始利用物理模拟来尝试预测蛋白质的折叠形态。此后,他们采用了各种各样的技术,如动力学分析、热力学研究和进化研究等。尽管这些技术长期以来进展缓慢,但即使在取得进展的情况下,也不免有人怀疑科学家们是否在有意挑选数据。比如,约翰霍普金斯大学在 20 世纪 90 年代曾声称基本上已经“解决”了蛋白质结构预测的问题。
为了公平且严格地评估科学进展,1994 年起,一个名为 CASP(蛋白质结构预测的关键评估)的比赛应运而生。CASP 每两年举办一次,要求参赛者对蛋白质结构进行盲目预测。具体来说,就是让他们预测那些已知氨基酸序列但三维结构尚未实验确定的蛋白质。生物学家们正在研究这些蛋白质的结构,并希望在比赛结束后不久就能确定,以此作为评分预测的依据。每个模型的评分大致基于正确预测氨基酸位置的百分比,且需要满足一定的严格标准6。从 2010 年代的数据来看,赢家的得分通常在 30 至 50 之间7:
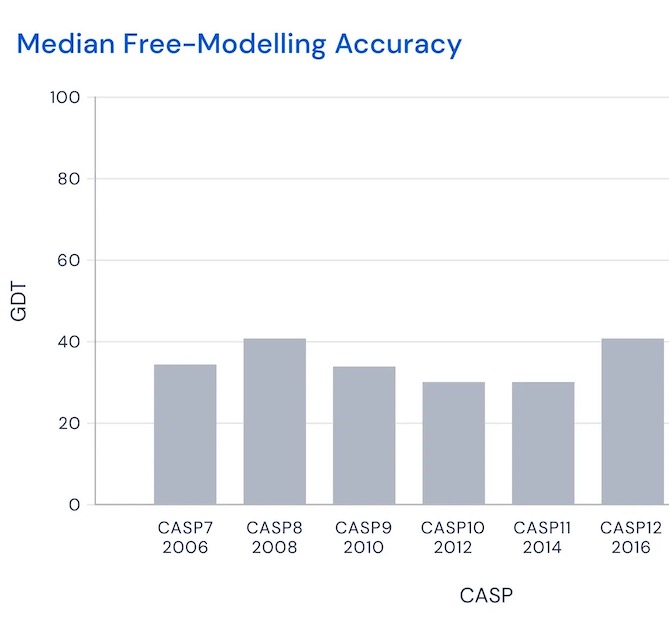
也就是说,获胜者大致能将一半以上的氨基酸非常准确地定位。然而在 2018 年和 2020 年,DeepMind 分别以 AlphaFold 和 AlphaFold 2 参加比赛,成绩显著:
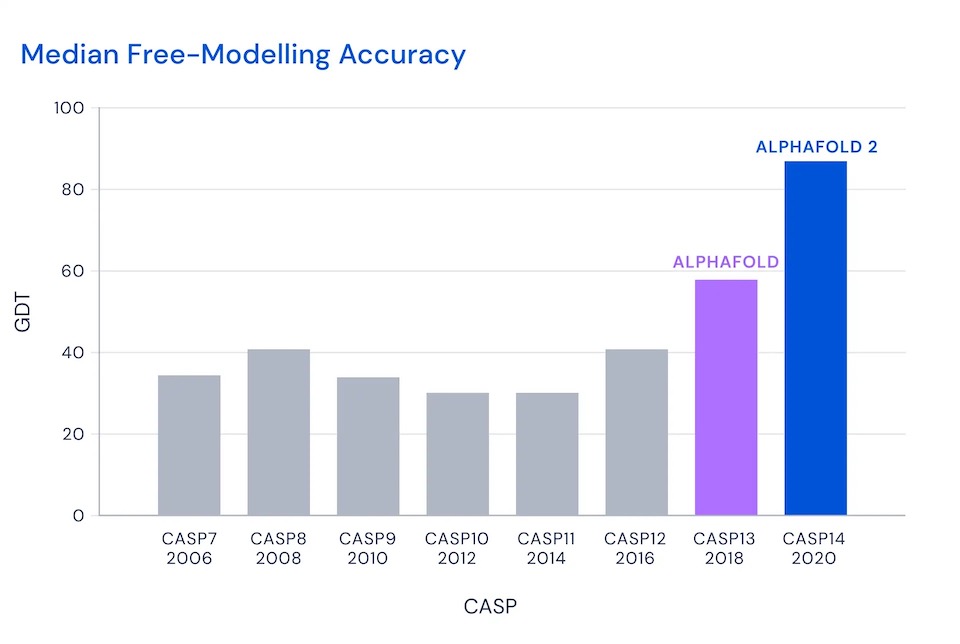
特别是在 2020 年的 CASP 14 比赛中,AlphaFold 2 取得了 87 分的高分,意味着大约 87% 的氨基酸在严格标准下被准确预测。这一分数是 AlphaFold 在最具挑战性的“自由建模”类别中取得的,该类别要求参赛者预测那些鲜为人知的蛋白质结构。在所有类别中,AlphaFold 预测的主链阿尔法碳原子的平均位置偏差仅为 0.96 埃8。相比之下,排名第二的技术准确度大约只有三分之一,其平均位置偏差为 2.8 埃9:
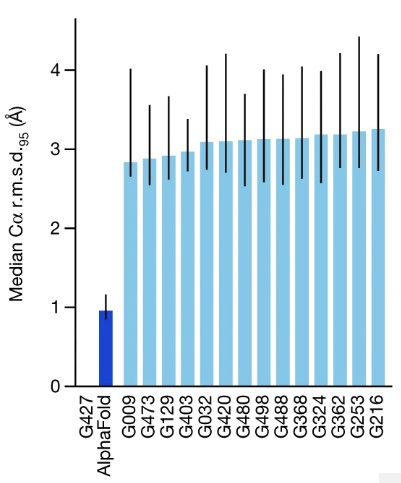
作为对比,碳原子的范德华直径大约为 1.4 埃。AlphaFold 的准确度通常可以与实验所得的准确度媲美。CASP 联合创始人、马里兰大学的 John Moult 曾说10:
我们几乎被蛋白质折叠这个难题困扰了 50 年。见证 DeepMind 提出解决方案,回想我个人长期致力于这个问题,经历了无数挫折与起伏,曾一度怀疑我们是否能找到答案,这真是一个极为特别的时刻。
这番话极具分量,值得深入探究 AlphaFold 在解决蛋白质结构预测问题上的意义,以及还有哪些问题待解。然而,即便是 AlphaFold 的竞争者也对其赞不绝口。哥伦比亚大学的 Mohammed AlQuraishi 这样说11:
这是否意味着解决了静态蛋白质结构预测的问题?我认为是的,尽管还存在一些微妙之处。在这里,有见地且深思熟虑的人可能会有不同观点,这取决于个人对“解决方案”这个词的理解……大部分科学难题已被解决;现在剩下的主要是实践操作。
那是两年半前的事了。现在一个普遍的观点是:AlphaFold 已经取得了巨大的进步,但即使在基本问题上仍有许多工作要做,许多新的巨大挑战现在也已成为可能的攻击目标。
AlphaFold 的高级架构复杂到需要几个小时才能完全描述。今天,我只想强调几个关键点:
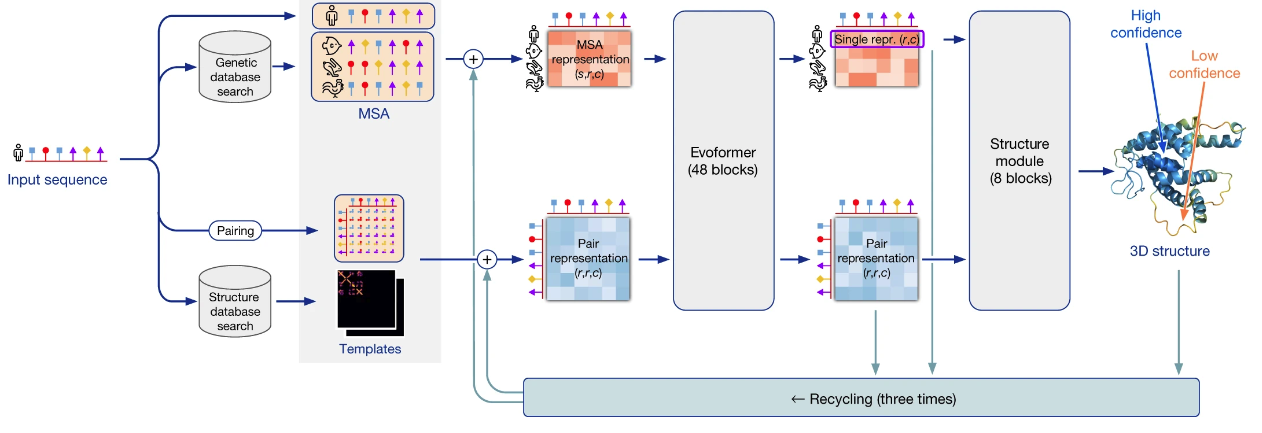
它是一个深度神经网络,即一个分层模型,通过训练学习了 9300 万参数。它接受氨基酸序列作为输入,输出一个三维结构和一些误差估计,后者用于量化 AlphaFold 对每个氨基酸位置的置信度。它的基本训练数据来自蛋白质数据银行(PDB),这是人类自 20 世纪 70 年代以来通过实验确定的蛋白质结构的集合。在训练阶段,共有 170,000 个蛋白质结构(尽管因技术原因省略了一小部分)。通过梯度下降方法调整 AlphaFold 网络中的参数,确保网络在给定输入的情况下输出正确的结构。
这是一个对 AlphaFold 学习预测蛋白质结构方法的宽泛概述。其实,还有许多其他重要的概念。其中一个巧妙而重要的已有想法是通过分析数亿已知的蛋白质序列来学习。这个方法的核心是寻找许多氨基酸序列与输入蛋白质相似的其他蛋白质,这些蛋白质可能与输入蛋白质在进化上有关。比如,它们可能在本质上是相同的蛋白质,但存在于不同的物种中。AlphaFold 试图找到这些相似蛋白质,并从中学习。为了更好地理解其方法,假设 AlphaFold 在观察许多相似蛋白质时发现,有一对氨基酸在线性链上相距较远,但其中一个氨基酸的变化似乎与另一个的变化相关联。这可能意味着这些氨基酸在空间上彼此靠近,并共同进化以维持蛋白质的结构。实际上,这种情况经常发生。它提供了一种方法,让 AlphaFold 既可以从蛋白质数据库中的已知结构信息(例如 PDB)学习,也可以从已知进化信息中学习。John Jumper 在一次演讲中精彩地阐述了这一点,他是 AlphaFold 论文的主要作者。他指出,物理学知识有助于 AlphaFold 理解进化史,而进化史又反过来指导 AlphaFold 理解物理学知识。
泛化与可靠性
你可能好奇:AlphaFold 是仅仅记住了它的训练数据,还是具备了泛化能力呢?CASP(蛋白质结构预测大赛)提供了一个基础的验证:在 AlphaFold 预测这些结构时,这些比赛中的结构并不在 PDB(蛋白质数据银行)中。而且,CASP 在某种程度上可以看作是一个“自然”的样本集:结构生物学界倾向于研究对广大生物学家来说重要的结构12。这表明 AlphaFold 或许能泛化到生物学家普遍感兴趣的蛋白质上。
如果我们利用深度学习研究自然界中不存在的蛋白质,比如由于突变产生的,或是蛋白质设计的一部分,会怎样呢?目前有大量的研究正在进行,现在的情况可以说是令人兴奋而又有些混乱。关于这个话题的概述至少需要覆盖几十甚至上百篇论文。这些论文中,有的指出 AlphaFold 在某些类型的突变上表现不佳,有的则表示在另一些类型的突变上表现良好,还有很多关于蛋白质设计的深度学习解决方案等等。我的看法是:生物学家将花费多年时间来研究这些不足之处,并改进系统以弥补这些不足。
OpenFold13 对深度学习泛化能力进行了一些有趣的测试,它是一个开源的、与 AlphaFold 非常相似的项目。比如,他们重新训练了 OpenFold,但从训练集中去掉了大多数的广泛拓扑类别。他们采用了 CATH 分类(一种蛋白质拓扑的标准分类方案),移除了训练集中高达 95% 的拓扑,然后从头开始重新训练 OpenFold。即便移除了大多数拓扑,OpenFold 的性能仍与 AlphaFold 114 相似:就在 AlphaFold 2 出现前的几年,它已经是最先进的技术了。
在这个理念的另一种尝试中,研究人员使用更小的蛋白质数据样本来重新训练 OpenFold。他们并非使用 170,000 个结构,而是采用了小至 1,000 个结构的训练集,这些结构是随机选取的。令人惊讶的是,即便是这样微小的训练集,他们也取得了与 AlphaFold 1 相媲美乃至略胜一筹的成效。而仅用 10,000 个结构作为训练数据,他们就实现了与完整版 OpenFold 相似的性能。
从实际应用的角度来看,AlphaFold 的泛化能力之所以重要,部分原因在于 DeepMind 发布了 AlphaFold DB。这是一个庞大的数据库,包含了 2.15 亿个蛋白质结构,涵盖了 48 种物种的几乎完整蛋白质组,包括人类、小鼠和果蝇等。这些结构是通过分析 UniProt 中的遗传序列,再应用 AlphaFold 预测结构得到的。整个过程可以概括为:
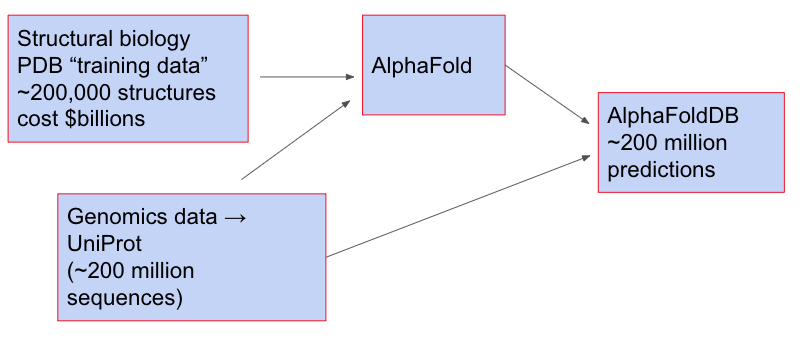
这是一次令人赞叹的泛化实践。如果我们拥有一个完全可靠的模型,那么在蛋白质结构的理解上,我们的认知将会扩展至少三个数量级。需要特别指出的是,AlphaFold 并未进行任何新的实验,也没有收集额外的数据。然而,仅凭理论推导,他们就能够得到大量的、被广泛认为是高质量的预测。我曾听到不少生物学家这样说:“虽然没有人会仅凭 AlphaFold 的预测就断定其准确性,但它无疑是一个极好的起点,能够为研究者节省数月的工作时间”。
随着模型的不断完善,我预见模型和实验之间的分界将逐渐模糊。这听起来可能有些奇怪,但其实传统的“实验确定结构”在将数据转化为结构时,实际上需要依赖复杂的理论。如果你更相信 AlphaFold(或其后续版本)提供的强大理论,你可能会更信任深度学习系统的预测,而不是现今的“实验结果”。这种现象已经有初步迹象。在 CASP 评估中,AlphaFold 在几个使用核磁共振 (NMR) 光谱学确定的结构上表现不佳。2022 年的一篇论文15研究了“904 个同时具有 Alpha-Fold 和 NMR 结构的人类蛋白”,并得出结论:“Alpha-Fold 的预测通常比 NMR 结构更准确”。论文作者之一,Mike Williamson,正是利用 NMR 进行结构生物学研究的先驱。
举个更简单的例子:我们对望远镜图像的解释取决于我们的光学理论;如果我们改变或改进光学理论,我们对望远镜“原始数据”的理解也会相应改变。实际上,我们对光线在引力作用下的弯曲理解已发生改变。在这种情况下,我们改变了对光通过空间的运行方式的理解,这影响了我们对实验数据的解释,尤其是在理解遥远星系的引力透镜效应等现象。
类似地,“实验性”蛋白质结构测定也极度依赖于理论。在 X 射线晶体学(一种蛋白质结构分析方法)中就能看到这一点,它需要多个复杂步骤:蛋白质样品纯化、蛋白质结晶、使用 X 射线衍射获得二维图像、反演并求解三维结构、以及判断反演是否足够好的标准。这一过程涉及大量理论!实际上,反演过程通常从一个好的“猜测”,即一个候选搜索结构开始。人们通常使用相关蛋白质作为搜索结构,但有时无法找到合适的搜索结构,这可能阻碍解决方案的找到。AlphaFold 已被用来寻找良好的搜索结构,以帮助反演特别具挑战性的数据。因此,理论与实验之间已经存在一条模糊的界线。我预计,在未来几年内,如何验证 AI“解决方案”将成为科学和元科学领域的一个重要议题。
能否简单总结蛋白质结构的原则?人工智能会助力我们的发现吗?
一个包含 9300 万学习参数的模型无疑是复杂的,并不像传统的“理论”或“解释”那样直观。你可能好奇:AlphaFold(或其后续产品)能否助力我们揭示这样的理论,哪怕是部分的?比如,是否存在一套简单的蛋白质结构预测原则?AlphaFold 2 到底学到了什么?先说一下,对于这些问题,我们目前还没有答案。但探索这些问题对于理解 AI 在科学中的作用极为重要。
一种发现这些原则的方法是“行为主义人工心理学”,即通过观察系统行为来推导出高层次原则。显然,AlphaFold 的详细预测已被生物学家广泛应用,例如用于发现蛋白质的新结合位点。这虽然非常有益,但并不等同于推理出关于蛋白质结构的全新高级原则。不过,在观察其他重要深度学习系统的行为时,人们已经发现了一些有趣的新高层原则。
例如,通过观察 AlphaZero 国际象棋系统的行为,人们推断出了一些违背传统国际象棋大师智慧的行为。这些发现分别在 2018 年 12 月17和 2019 年 1 月18公布。一项最新研究19分析了这些行为,并探究了人类棋手是否因此系统而改变了自己的棋风。研究发现,2019 年世界前十棋手中几乎无人改变,唯一的例外是当时和现在的世界顶尖棋手,Magnus Carlsen。他们指出 Carlsen 在 2019 年的棋风中发生了显著变化,这很可能受到了 AlphaZero 的影响。
在这些变化中,Carlsen 在比赛的早期阶段显著增加了 h 线兵的推进频率,使用白棋时增加了 333%,使用黑棋时增加了 175%【20】。他针对白棋和黑棋均调整了开局策略。实际上,他在 2019 年最常用的两种白棋开局(女王弃兵拒绝和格鲁菲尔德防御的变体)是他在 2018 年完全未曾使用的。另外,他在 2018 年使用黑棋时不到 10% 的情况下选择西西里防御,但到了 2019 年这一比例上升至 45%。他们还观察到 Carlsen 在牺牲棋子方面变得更加愿意。所有这些变化都显示出他从 AlphaZero(AlphaZero)那里学到了很多。Carlsen 在 2019 年没有输掉任何传统棋局,并且是唯一一位显著提升 Elo 等级分(Elo rating)的顶尖棋手,尽管他已是排名最高的棋手:他的等级分提高了 37 分。相比之下,前十名棋手中有六人失分,其余三人没有一人增加超过 6 分。当然,我们不清楚这些成就有多少是因为 Carlsen 从 AlphaZero 学习的结果,但这篇论文为 Carlsen 从 AlphaZero 学到许多做出了合理的论证。
这种行为观察虽有趣,但也让人感到挫折,因为它并没有解释这些行为背后的原因。我们或许知道 AlphaZero 喜欢在早期推进 h 线兵,但更重要的是理解这背后的原因。我们能否深入神经网络内部,理解它们是如何实现这些行为的?据我所知,针对 AlphaFold(AlphaFold)的这类调查只是偶尔进行。但对于更简单的神经网络,人们已经发现了一些有趣的结构。比如去年,Neel Nanda 和 Tom Lieberum【21】训练了一个单层 Transformer 神经网络,用以实现两个整数模 113 的加法。最初,这个网络仅仅记住了训练集中的所有例子,它能够很好地完成这些特定例子的加法,但在其他情况下表现很差。然而,随着训练时间的延长,网络在未出现在训练集中的例子上的表现大幅提升。不可思议的是,它在没有额外训练数据的情况下,开始学会了加法运算。
Nanda 和 Lieberum 花费数周时间深入探究了网络内部发生的变化。他们通过逆向工程的方法发现,网络以一种非常特别的方式学会了加法。大体上,对于给定的数字 和 ,网络计算出一个波形 ,接着进行相位转换至 ,最终寻找一个能抵消相位移动的 ,使得波形 尽可能地“平稳”。这种做法就像是无线电频率工程师或群表示理论家处理加法的方式。这对 Nanda 和 Lieberum 来说是个巨大的惊喜。Nanda 这样说道:
我要强调,这个算法完全是通过梯度下降(gradient descent)学习到的!我事先并没有预料到或理解这种算法,也没有采取任何措施去引导模型学习这种模块化加法的方式。这一发现完全是通过逆向工程模型的权重而得到的。
当我第一次听到这个故事时,我感到非常困惑:为什么网络会转向这种更通用的算法呢?毕竟,它最初只是记忆了训练数据,并且表现出色:为什么要发生变化呢?答案在于训练过程中,神经网络会因为采用更复杂的模型而付出代价:损失函数是这样设计的,使得梯度下降会偏好权重较低的模型。波形算法实际上就是一种权重较低的算法。这其实是奥卡姆剃刀原则(Occam's razor)的一种实践,即倾向于选择更简单、更通用的方法。实际上,通过调整损失函数,我们可以用许多不同的方式来应用奥卡姆剃刀原则。这让人深思:有朝一日,我们是否也能对像 AlphaFold 这样的系统做出类似的发现,揭示出蛋白质结构的新基本原理呢?
思考总结
我希望你们喜欢这篇关于 AlphaFold 的简介和对 AI 在科研领域应用的思考。实际上,关于这个话题还有很多值得探讨的内容。但显而易见的是,AI 系统引发了众多关于基础科学和元科学的重要问题。最关键的是:随着它们在各种认知任务中的能力日益增强,以及在环境中的感知和作用能力不断提升,它们将如何影响科学的整体发展?AI 系统是否会彻底改变科学研究的整个实践过程?它们能否显著加快科学发现的步伐?如果真的如此,这又将带来哪些风险和机遇?
后记:对于数据更复杂、不那么完善的问题,我们应该如何应对?
我的演讲结束后,观众提出了许多引人深思的问题。其中一个特别引人注目的问题是 Evan Miyazono 提出的。他的问题大致是:“蛋白质结构预测作为 AI 的一个目标,因为有大量清晰、详尽的先验数据可供学习而受益匪浅。那么,对于没有大量清晰先验数据的问题,AI 的作用有多大呢?”
这个问题非常好。如果是一年前,我可能会说,在处理数据更杂乱、成功指标不明确的问题时,应用 AI 确实挑战重重。但在过去的一年中,随着我对基础模型(foundation models)、迁移学习(transfer learning)和零样本学习(zero-shot learning)有了更深入的理解,我的看法发生了改变。
让我举两个例子来说明为什么我会改变看法。首先,来说说一些非官方的小道消息:很多人发现 ChatGPT 在编程方面比 GitHub Copilot 更为出色。具体原因我不太清楚,但根据我从业内人士那里听到的一些推测,训练模型处理大量文本(包括代码)能显著提升它的编程能力。看来,文本中的规律性在某种程度上增强了 ChatGPT 理解代码的能力。
第二个例子是关于 AlphaFold。要说明这一点,我需要补充一下我在演讲主体中未提及的 AlphaFold 训练细节:它实际上包含了一个大语言模型,把氨基酸序列当作一种待填充的“文本”。具体来说,AlphaFold 会根据给定的氨基酸序列,在已有的遗传数据库中寻找相似序列。在训练过程中,就像语言模型一样,AlphaFold 会掩盖(或变异)某些氨基酸,并尝试预测被遮蔽的部分。这种做法迫使网络更深入地学习进化信息的结构,进而利用这种深入理解来更准确地预测蛋白质结构。这样,AlphaFold 在获得遗传学领域的大量信息的同时,也从这个相关领域的迁移学习中受益。
在科学的广阔背景下,我预测大型多模态基础模型将逐步超越单一用途的系统。这种趋势类似于大语言模型(大语言模型)通常超过更专门的自然语言处理算法。这些模型还将展现零样本(零样本)或少样本(少样本)学习的能力。这些大型多模态模型将不仅在文本上进行训练,还会在图像、动作、代码、遗传数据、地理数据以及各种传感器和执行器数据上进行训练。与语言模型相似,它们将利用在某一领域的理解,来辅助处理其他领域的问题,尤其是那些信息不完整、更为复杂的领域。事实上,蛋白质语言模型已经显示出朝这个方向发展的迹象。
当然,这些目前还只是我的想象和直觉。这一切还未成为现实,除了一些非常初步的尝试。我可能完全错误!但我确信,要让这一切顺利进行,需要新的思路和方法。然而,我认为,未来几年内,AI 模型会在处理只有少量先前数据的复杂问题方面表现出惊人的能力。它们将依靠多模态基础模型、迁移学习(transfer learning)以及零样本和少样本学习来实现这一点。
致谢
这次演讲得到了 Astera Institute 的支持。感谢 Alexander Berger、David Chapman、Evan Miyazono、Neel Nanda 和 Pranav Shah 的宝贵意见,这些意见帮助我改进了这次演讲。同时,也感谢 Twitter 上的许多朋友,他们帮助我深入理解 AI 和分子生物学。
引用信息
在学术环境中引用此项工作,请引用为:
Michael Nielsen, "How AI is impacting science", https://michaelnotebook.com/mc2023/index.html, 旧金山 (2023)。
脚注
-
John Jumper, Richard Evans, Alexander Pritzel 等人, 通过 AlphaFold 实现的高精度蛋白质结构预测, Nature (2021)。另请参阅 这个 colab,它提供了 Google DeepMind 开发的 AlphaFold 的可操作版本,这是 AlphaFold 背后的公司。请注意,其他源头提供了更快速的相似性能的实现方式,例如此 ColabFold colab。我们稍后将看到,其他团队也开发了用于训练该系统的开源代码,不仅限于做结构预测。↩︎
-
在本文中,我将用 "AlphaFold" 专指 AlphaFold 2。显然,它是继早期的 AlphaFold 蛋白质结构预测系统之后的版本。但我们不会深入探讨早期系统,因此我通常会省略数字标识。值得注意的是,AlphaFold 1 和 AlphaFold 2 在架构上有很大差异,尽管它们都是蛋白质结构预测的深度学习系统。↩︎
-
事实上,在宏基因组学数据库中可能会发现数以十亿计的数据(虽然这些数据的来源不甚明确)。↩︎
-
这是一个复杂的话题,有时被称为 Anfinsen 的假设或 Anfinsen 的教条。这个假设并非在所有情况下都成立 — 当然存在着一些特定情况,其中相同的氨基酸序列会以不同的方式折叠,或者根本没有稳定的结构。然而,它作为理解蛋白质及其功能的起点,其实用性极高。在这次讲话中,我大体上会假定这一假设是正确的。↩︎
-
举个例子,约翰斯·霍普金斯大学发布了一篇引人注目的新闻稿,介绍了 George Rose 和 Rajgopal Srinivasan 在预测球形蛋白质主链结构方面取得的重要进展。↩︎
-
具体来说,CASP 中用到的全球距离测试(GDT)是基于四个不同的比率计算出的平均值,这些比率反映了预测的主链 alpha 碳原子与实验结构相比,在 1, 2, 4, 和 8 埃距离内的准确度。作为参考,通常认为高质量的实验结构的精确度在大约 1-2 埃左右;虽然有时可以做得更好,但更差的结果也时有发生,这取决于使用的技术。↩︎
-
这张图表及其后续的图表是基于AlphaFold: 生物学中 50 年大挑战的解决方案(2021)的数据改编而成的。↩︎
-
这项数据是在 alpha 碳原子 95% 覆盖率下得出的。若达到 100% 覆盖率,则精确度为 1.5 埃。↩︎
-
该图取自 John Jumper, Richard Evans, Alexander Pritzel 等人的研究,题为《AlphaFold:高精度蛋白质结构预测》, 发表于《自然》杂志(2021)。↩︎
-
引述自:AlphaFold: 生物学中 50 年大挑战的解决方案(2021)。↩︎
-
Mohammed AlQuraishi, AlphaFold2 @ CASP14: “它就像是我亲手抚养长大的孩子,现在已经离家远去。” (2020).↩︎
-
当然,像实验结构的可行性这类其他因素在研究中也扮演着不可或缺的角色。↩︎
-
Gustaf Ahdritz, Nazim Bouatta, Sachin Kadyan 等人,OpenFold: 重新训练 AlphaFold2 揭示其学习机制和泛化能力的新洞见 (2022).↩︎
-
他们采用了本地距离差异测试(lDDT-C)作为评估标准,而不是 AlphaFold 在 CASP 中所强调的全球距离测试。我对这种选择是否真的有重大影响保持怀疑态度,但尚未进行验证;坦白说,我在思考是否有我未察觉的重要细节。↩︎
-
Nicholas J. Fowler 和 Mike P. Williamson, 通过 AlphaFold 和 NMR 确定溶液中蛋白质结构的准确性, Structure (2022).↩︎
-
David Silver, Thomas Hubert, Julian Schrittwieser 等人,通过自我对弈和通用强化学习算法掌握国际象棋和将棋 (2017).↩︎
-
David Silver, Thomas Hubert, Julian Schrittwieser 等人,"一种能够通过自我对弈精通国际象棋、将棋和围棋的通用强化学习算法", Science (2018)。↩︎
-
Natasha Regan 和 Matthew Sadler, "游戏规则的颠覆者:AlphaZero 颠覆传统国际象棋策略及其对人工智能的深远影响" (2019)。↩︎
-
Julio González-Díaz 和 Ignacio Palacios-Huerta, AlphaZero 的创新理念 (2022)。↩︎
-
这些研究都集中于传统的棋类游戏。↩︎
-
Neel Nanda 和 Tom Lieberum 最初进行了这项工作,并在 对“Grokking”(一种深度学习现象) 的机械解释性分析 (2022) 中进行了介绍。更全面的研究报告由 Neel Nanda, Lawrence Chan, Tom Lieberum, Jess Smith, 和 Jacob Steinhardt 在 通过机械解释性方法测量“Grokking”现象的进展 (2023) 中提供。↩︎